
Summary
Missense mutations in DNA, that lead to changes in amino acid content in proteins, are a common cause of several debilitating diseases. The challenge of identifying functionally crucial sites within proteins is underscored by the scarcity of comprehensive, and systematic datasets. The characterization of functional sites in proteins is often derived through multiple alignment methods that determine consensus/ or conserved amino acid sequences. However, conserved sequences are often essential for protein stability in solution, rather than functional or enzymatic activity.
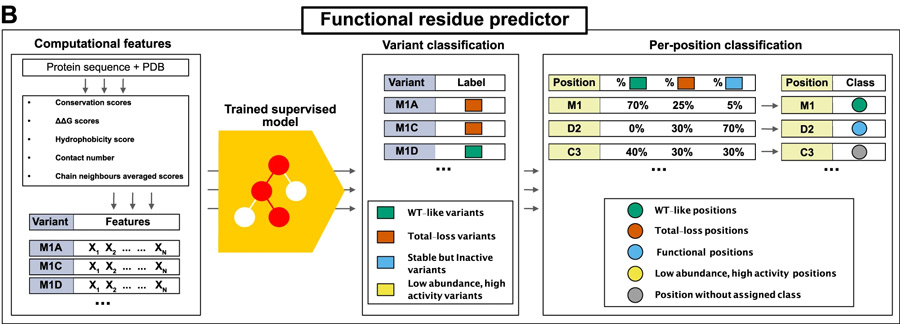
In response to these challenges, Cagiada et al. (2023) developed a sophisticated machine learning model, published in Nature, to predict functionally significant sites in proteins. Their methodology is a fusion of statistical models for protein sequences with biophysical models that gauge stability. This innovative model excels at identifying a spectrum of functional sites, encompassing active domains, as well as regulatory and binding sites. Researchers demonstrated the utility of their machine learning model by applying it to predict and subsequently experimental validate the repercussions of missense variants in HPRT1, which has been linked to Lesch-Nyhan syndrome. Through this, they gained profound insights into the molecular mechanisms underpinning the onset of this disease. This innovative approach promises to reshape the way we explore proteomics, thereby bringing us closer to unlocking their secrets and harnessing their potential for the betterment of science and medicine.
Introduction
Proteins are the workhorses of biology, involved in a myriad of crucial functions, from catalyzing chemical reactions to transmitting signals within cells. Understanding the functional sites within proteins that are important for their activity is of paramount importance in the fields of molecular biology, biotechnology, and pharmacology. After all, these sites hold the key to unraveling the mysteries of diseases, aiding in the development of personalized therapeutic treatments, and engineering proteins for specific purposes. But how do we identify these critical sites in proteins?
The Complexity of Protein Function
Proteins have specific sites or regions within their structure that are responsible for their biological function (e.g., active sites, protein-protein interfaces, allosteric sites, and regions that interact with substrates or ligands). Identifying the active/ or functional sites in proteins, however, is not a straightforward task. Many amino acid substitutions that impact protein function also affect stability and cellular abundance, making it challenging to distinguish the direct functional effects from structural stability changes.
A Machine Learning Solution
To address the challenge of identifying functional sites in proteins, a state-of-the-art machine learning method was developed, which combines statistical models for protein sequences with biophysical models of stability. This model is trained using experimental data on variant effects and validated extensively. It offers the capability to predict a wide range of functional sites, including active sites and regulatory sites, with remarkable accuracy.
Prospective Prediction and Experimental Validation
The true power of this model lies in its ability to predict the functional consequences of missense variants in proteins. For instance, it can be used to pinpoint the molecular mechanisms by which variants in protein coding sequences cause diseases like Lesch-Nyhan syndrome. This predictive capability can be a game-changer in healthcare research, and the development of personalized therapeutic treatments.
Making the Model Accessible
Fortunately, the creators of this predictive machine learning model have made it accessible to the entire scientific community in the elegant format of a notebook that guides users through the process of generating input data and making predictions of functional sites. Being able to run this model via Google Colaboratory not only makes the technology available but also encourages collaboration and further research in the field. This type of open accessibility should be celebrated because it increases the liklihood that more scientific minds can work with such a powerful predicitve model!
Conclusion
Understanding the functionally important sites in proteins is a cornerstone of advancing our knowledge of biology and disease. This machine learning model is poised to accelerate our progress in these areas. Whether it’s selecting residues for protein engineering experiments, generating focused libraries in enzyme optimization, or unraveling the molecular basis of diseases, this technology opens up new frontiers for research and discovery.
Outsourcing Bioinformatics Analysis: How Bridge Informatics Can Help
Groundbreaking studies like these are made possible by technological advances making biological data generation, storage, and analysis faster and more accessible than ever before. From pipeline development and software engineering to deploying existing bioinformatics tools, Bridge Informatics can help you on every step of your research journey.
As experts across data types from leading sequencing platforms, we can help you tackle the challenging computational tasks of storing, analyzing and interpreting genomic and transcriptomic data. Bridge Informatics’ bioinformaticians are trained bench biologists, so they understand the biological questions driving your computational analysis. Click here to schedule a free introductory call with a member of our team.
Figure 1 – source: https://www.nature.com/articles/s41467-023-39909-0#Fig1
Dan Ryder, MPH, PhD
Dan is the founder and CEO of Bridge Informatics, a professional services firm helping pharmaceutical companies translate genomic data into medicine. Unlike any other data analytics firm, Bridge forges sustainable communication change between their client’s biological and computational scientists. Dan is particularly passionate about improving communication between people of different scientific backgrounds, enabling bioinformaticians and software engineers to collectively succeed.
Prior to forming Bridge Informatics, Dan served in a variety of roles helping pharmaceutical clients solve early-phase drug discovery and development challenges.
Dan received both a Ph.D. in Biochemistry and Molecular Biology and an MPH in Disease Control from the University of Texas Health Science Center at Houston (UTHealth Houston). He completed his postdoctoral studies in Molecular Pathways of Energy Metabolism at the University of Florida College of Medicine. Dan received his undergraduate degree in Microbiology from the University of Texas at Austin.